最近发布



清华大学王定胜教授联合山东大学张进涛教授合作利用RapidXAFS表征铁镍分子配合物OER催化剂发表在JACS

全文速览
具有活性单位点的分子配合物在电催化转化过程中展现出巨大潜力。然而,配合物与碳载体之间的空间间隔对电子穿梭的影响仍鲜有研究。针对此问题,清华大学王定胜课题组联合山东大学张进涛团队,提出了一种超分子构筑策略,利用氧官能团修饰碳载体,强化配合物与载体之间的相互作用,从而阐明空间间隔对电催化析氧反应(OER)机制的影响。实验结果表明,较小的空间间隔有利于电子穿梭并稳定分子配合物,从而实现安培级的电流密度。优化后的联苯-4,4'-二甲酸配位的FeNi配合物表现出卓越的OER性能,在过电位为0.33 V 时,电流密度可达1.5 A cm-2,质量活性高达41,206 A gFe/Ni-1。理论研究进一步表明,Fe和Ni位点之间形成的高度亲电氧桥会促进铁位点上关键中间体(Fe-OOH*)的形成。这些发现表明,配合物与载体之间的相互作用大小对异相分子配合物的本征活性起着重要作用。

背景介绍
由可再生能源驱动的OER在各种电化学能量转换反应中被视为一种极具前景的绿色途径。由于其多步骤的质子耦合电子转移过程,要获得足够高的电流密度就需要消耗大量能量,从而降低了催化效率。近年来,有机-金属杂化催化剂,如MOFs、COFs以及配位聚合物,由于其可调配位环境、原子分散的活性位点以及协同的金属-配体相互作用,被用来解决缓慢的 OER 速率问题。然而,大多数金属位点都深深地嵌入材料内部,难以接触到电解质。这导致了材料重建困难以及电子从电极到催化剂的传输受限,使得电流密度低于安培级别。碳载体负载的具有高度暴露的金属中心、功能性的配体以及可调的配位环境的均相分子配合物已被大量用于电催化还原过程,能够实现 > 100 mA cm-2的电流密度。然而,用于OER的这类分子配合物却鲜有报道。主要挑战在于分子配合物与导电碳基底之间的巨大空间间距,这是由于弱的物理吸附所致。这限制了电子转移并减缓了反应动力学,从而限制了电流密度。此外,复合物与支持之间的弱相互作用可能导致材料整体的不稳定。因此,迫切需要构建异相分子配合物,以在安培级电流密度下稳定地催化OER,这凸显了调节配合物-载体相互作用以及催化剂稳定性方面进一步创新的必要性。
本文亮点
1. 氧官能团修饰策略调节配合物-载体相互作用
通过浓硝酸处理科琴黑,在碳载体上引入吸电子氧基团,在配合物和载体之间形成强电子偶联 ,制备了氧化科琴黑。然后在溶液中通过金属离子和联苯二甲酸的快速组装,形成了碳载体负载的金属-有机分子配合物(FeNi-BPDA)。表征结果表明,氧化科琴黑负载的FeNi-BPDA(FeNi-BPDA/C(O))相比于科琴黑负载的FeNi-BPDA(FeNi-BPDA/C)具有更窄的空间间距,这有利于分子配合物与载体之间的电子穿梭,为实现高OER性能奠定了基础。
2. 卓越的催化活性
FeNi-BPDA/C(O)呈现出卓越的安培级OER催化性能,在过电位为0.33 V时,电流密度可达1.5 A cm-2,质量活性高达41,206 A gFe/Ni-1,远超于未经氧官能化处理的碳载体负载的分子催化剂FeNi-BPDA/C。在阴离子交换膜电解槽中展现出了优异的催化效率和大电流密度下的催化耐久性。
3. 反应机制探究
原位光谱表征和XAS谱证明,分子配合物与碳载体之间缩小的间距能够增强电子转移过程,从而有助于实现高氧化态Fe-O2-Ni位点的转变。理论研究表明,生成的Fe-O2-Ni位点具有高度亲电性的氧桥,能够通过OH-亲核攻击形成Fe-OOH*中间体,从而实现安培级电流密度。
图文解析
 图1 FeNi-BPDA/C(O)和FeNi-BPDA/C的结构设计与表征
图1 FeNi-BPDA/C(O)和FeNi-BPDA/C的结构设计与表征
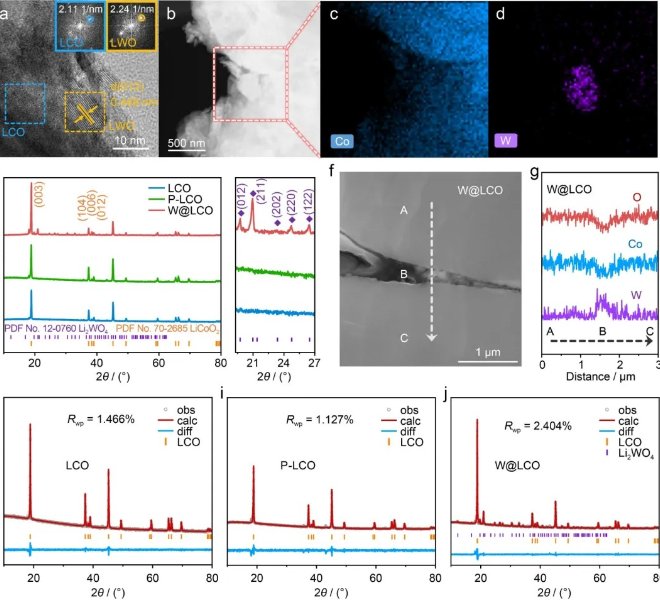
通过氧官能团修饰碳载体的方式,将FeNi-BPDA分子配合物与碳载体之间的空间间距缩小(图1a)。FT-IR光谱确认了含氧官能团掺入了科琴黑载体中(图1b)。XRD图证明FeNi-BPDA/C(O)和FeNi-BPDA/C中未形成MOF片层结构(图1c)。高分辨HAADF-STEM图像进一步证明了FeNi位点的原子及分散结构(图1d,e)。XPS结果证明FeNi-BPDA/C(O)相比于FeNi-BPDA/C,Fe和Ni位点均有价态的升高(图1f,g)。以上结果证明了材料的成功合成,且初步证明了FeNi-BPDA/C(O)中更有利的电子穿梭过程。
图2 XAFS表征
XAFS谱学技术是研究催化剂活性位点最有效的手段之一,本次实验部分XAFS数据由安徽吸收谱仪器设备有限公司的桌面X射线吸收谱仪测得,型号为RapidXAFS 2M。
FeNi-BPDA/C(O)和FeNi-BPDA/C在(a)Fe K边和(b)Ni K边的XANES光谱表明,FeNi-BPDA/C(O)具有更高的氧化态,这与XPS结果一致(图2a,b)。价态分析进一步精细说明了FeNi-BPDA/C(O)中FeNi位点略有升高的价态(图2c,d)。FT-EXAFS图及相应拟合结果表明,FeNi为原子及分散结构(图2e,f),且通过对空间间距(通过Fe–O₂/C₂和Ni–O₂/C₂键长表示)的统计可知,FeNi-BPDA/C(O)中分子配合物与载体之间的间距更窄(图2g)。该结果证明了FeNi-BPDA/C(O)和FeNi-BPDA/C中分子配合物的配位结构,并从实验角度给出了空间间距大小对比的证明。 
图3 电催化OER性能
通过不同催化剂在1.0 M KOH中测得的CV曲线可知,氧化科琴黑负载的分子配合物材料均有相比于科琴黑负载材料更大的氧化还原峰面积,证明更易发生的结构转变过程(图3a)。另外,氧化科琴黑负载的分子配合物材料也具有更高的单位总金属量活性(图3b),证明了窄的配合物-载体空间间距对材料整体性能的提升作用。LSV曲线证明了材料催化OER过程可达安培级电流密度(图3c),FeNi-BPDA/C(O)具有更高的反应速率(图3d)。在1.56 V电位下,FeNi-BPDA/C(O)具有很高的质量活性与单位总金属量活性(图3e)。另外,在200 mA cm⁻²下FeNi-BPDA/C(O)具有超40 h的催化稳定性(图3f)。OER过程中电解液中FeNi-BPDA/C(O)的Fe和Ni析出浓度更低(图3g),且稳定性测试后FeNi-BPDA/C(O)的高分辨HAADF-STEM图像仍基本保留原子级分散位点的形貌。以上结果说明了FeNi-BPDA/C(O)卓越的催化OER活性和稳定性。

图 4 OER过程中材料的动态结构转变
FeNi-BPDA/C(O)和FeNi-BPDA/C在不同电位下的ATR-SEIRAS光谱表明,反应过程中形成了金属-O*的中间体(图4a,b),且在FeNi-BPDA/C(O)中该结构更易形成(图4c)。不同电位下的原位拉曼光谱表明,FeNi-BPDA/C(O)具有更易发生的Ni-OOH转变过程(图4d,e)。接下来,FT-EXAFS拟合光谱表明,FeNi-BPDA/C(O)更易形成Fe-O-Ni结构(图4f,g),这也是真正的活性位点。另外, OER过程前后FeNi-BPDA/C(O)和FeNi-BPDA/C中Fe-O键长均出现明显的收缩(图4h),这表明Fe图 5 OER机制探究位点是被羟基亲核攻击发生含氧中间体转变的活性中心。
DFT模型构建表明,FeNi-BPDA/C(O)具有更窄的理论空间间距(图5a,b),这使得FeNi活性位点更易与载体之间发生电子穿梭,并使得材料整体具有更高的稳定性(图5c)。Bader电荷转移结果从理论角度证明了FeNi-BPDA/C(O)中FeNi位点具有更高的氧化态(图5d),这使得FeNi-BPDA/C(O)中Fe-O-Ni位点更易被羟基亲核攻击,且更易发生脱氢转变过程(图5e,f)。另外,FeNi-BPDA/C(O)进需要0.11 eV的理论过电位就可以驱动反应进行(图5g)。这些计算结果从理论层面阐明了FeNi-BPDA/C(O)的高活性来源,证明了配合物-载体相互作用调节对催化剂整体性能的调节作用,与实验观察高度一致。
总结与展望
在研究中,提出了一种超分子载体结构设计方法,该方法利用氧基团修饰碳载体来增强金属-有机分子配合物与载体之间的相互作用,从而有效地调节分子配合物与氧化碳载体之间的空间间距。具体来说,通过氧官能化碳载体负载了BPDA 配位的FeNi基分子配合物(FeNi-BPDA/C(O)),其在电催化水氧化OER过程中表现出出色的大电流密度催化性能,可达到1.5 A cm-2的电流密度,在过电位为 0.33 V 时质量活性高达41,206 A gFe/Ni-1,远超于其他已报道的金属-有机配合物催化剂。原位光谱表征和理论计算表明,分子配合物与载体之间的狭窄的间距有利于FeNi活性中心与碳载体之间高效的电子穿梭,从而通过连续的羟基亲核攻击和脱氢过程实现高价态Fe-O2-Ni位点的动态构筑。这些高度亲电性的氧桥通过O-O键与羟基偶联,以促进Fe-OOH*中间体的形成,进而实现快速的氧析出反应。我们的研究结果为高性能分子催化剂的设计提供了关键性的见解,即通过调控分子配合物与碳载体之间的空间间距来实现这一目的,这为开发下一代分子电催化剂提供了一种新的策略。